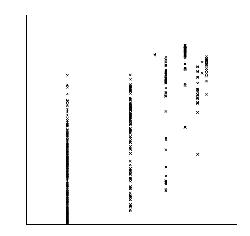
 Xdet implements two methods for detecting residues responsible for functional especificity in multiple sequence
alignments. These kind of positions, presenting a family-dependent (or function-dependent) conservation pattern, complement the fully-conserved positions as predictors of functionality. They are usually related to functional specificity.
Xdet implements two methods for detecting residues responsible for functional especificity in multiple sequence
alignments. These kind of positions, presenting a family-dependent (or function-dependent) conservation pattern, complement the fully-conserved positions as predictors of functionality. They are usually related to functional specificity.
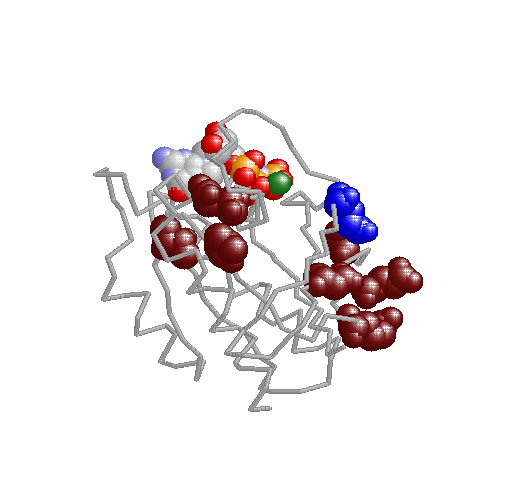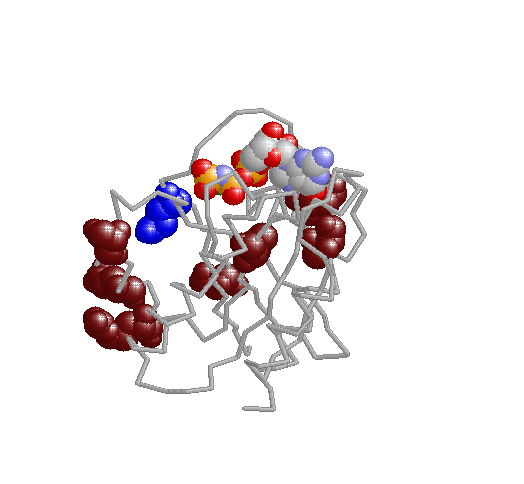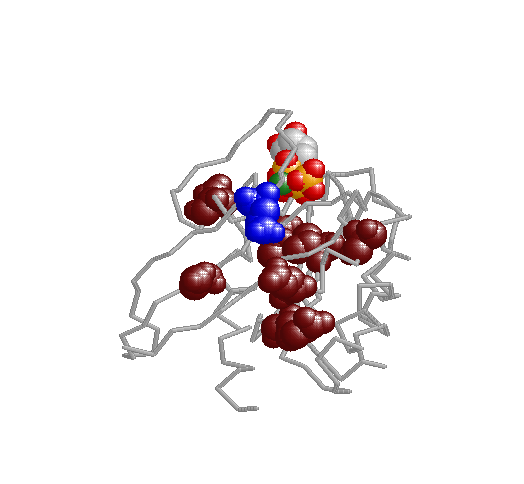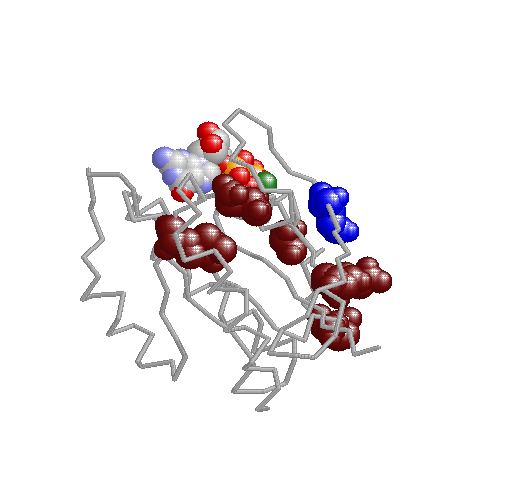
Xdet is now included in the JDet package. In that way, you can run Xdet and inspect its results in an interactive graphical user interface. If, for whatever reason, you want a single command-line version of Xdet not include in JDet's distribution, please contact Florencio pazos in the address below.
The documentation of the program, including information for using it and interpreting the results is available in this README file
Please, cite the references above when reporting any data obtained using this program.
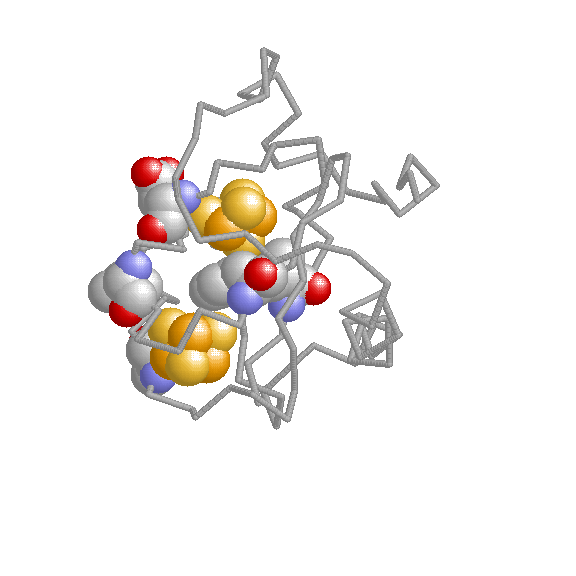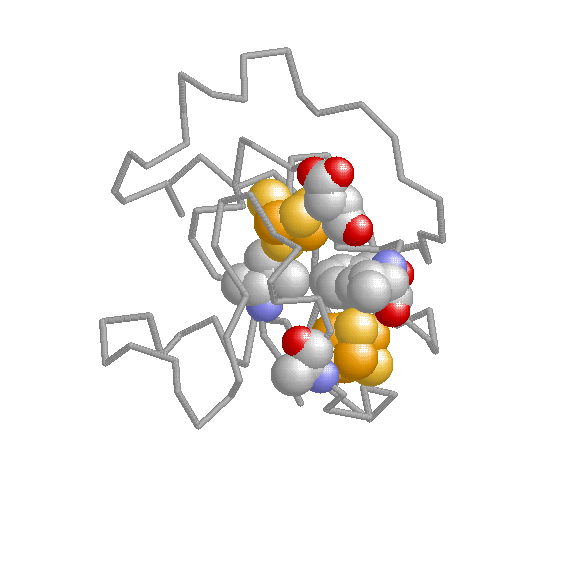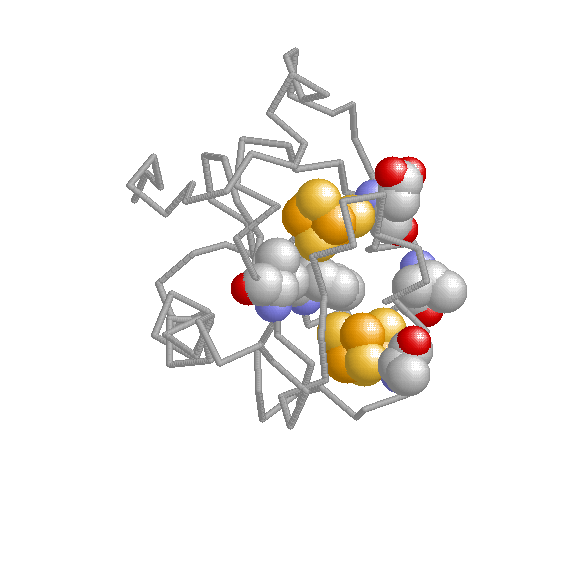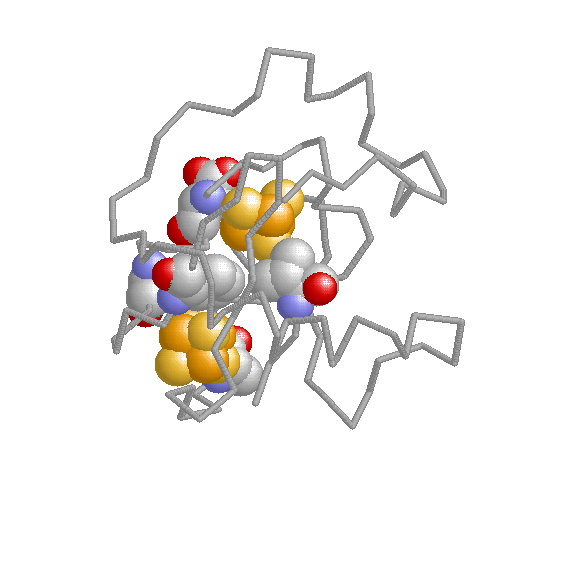 Additional resources:
Additional resources: